汕头大学钟建基团队开发了一种光氧化还原方法,实现了未活化烯烃与脂肪族羰基化合物的偶联,构建了一系列叔醇类化合物。将该光氧化还原策略应用于连接硼自由基反应,实现了烯烃与脂肪族羰基化合物的多向偶联反应。该反应可兼容多种烯烃偶联试剂,包括α-三氟甲基烯烃、脂肪族烯烃、杂原子取代烯烃、苯乙烯及缺电子烯烃,较现有方法取得显著突破。该方法具有多重关键优势:无金属参与且反应温和,对官能团具有优异兼容性,并可通过简单调整催化体系实现可调的化学选择性。
Abstract
The activation of aliphatic carbonyls to form ketyl radicals for C-C bond construction remains a formidable challenge, primarily due to their highly negative reduction potentials. Conventional strategies typically rely on stoichiometric metal reductants or strongly reducing conditions. Herein, we disclose a general photoredox platform that leverages ligated boryl radicals to efficiently activate aliphatic carbonyls, thus enabling divergent hydroxyalkylation of alkenes under metal-free and mild conditions. Notably, this strategy accommodates a wide range of alkenes, including α-trifluoromethyl olefins, aliphatic olefins, heteroatom-substituted alkenes, styrenes, and electron-deficient alkenes. Furthermore, by fine-tuning the catalytic system, the coupling of α-CF3 alkenes with aliphatic carbonyls can be selectively directed toward either defluoroalkylation or hydroalkylation products with high chemoselectivity, highlighting the versatility and tunability of this approach. Mechanistic studies confirm the critical involvement of ligated boryl radicals in the carbonyl activation. The synthetic utility of this protocol is further demonstrated through late-stage functionalization of pharmaceutically relevant molecules. With its broad substrate scope, good functional group compatibility, and programmable selectivity, this method provides a general and practical strategy for functionalization of aliphatic carbonyls.
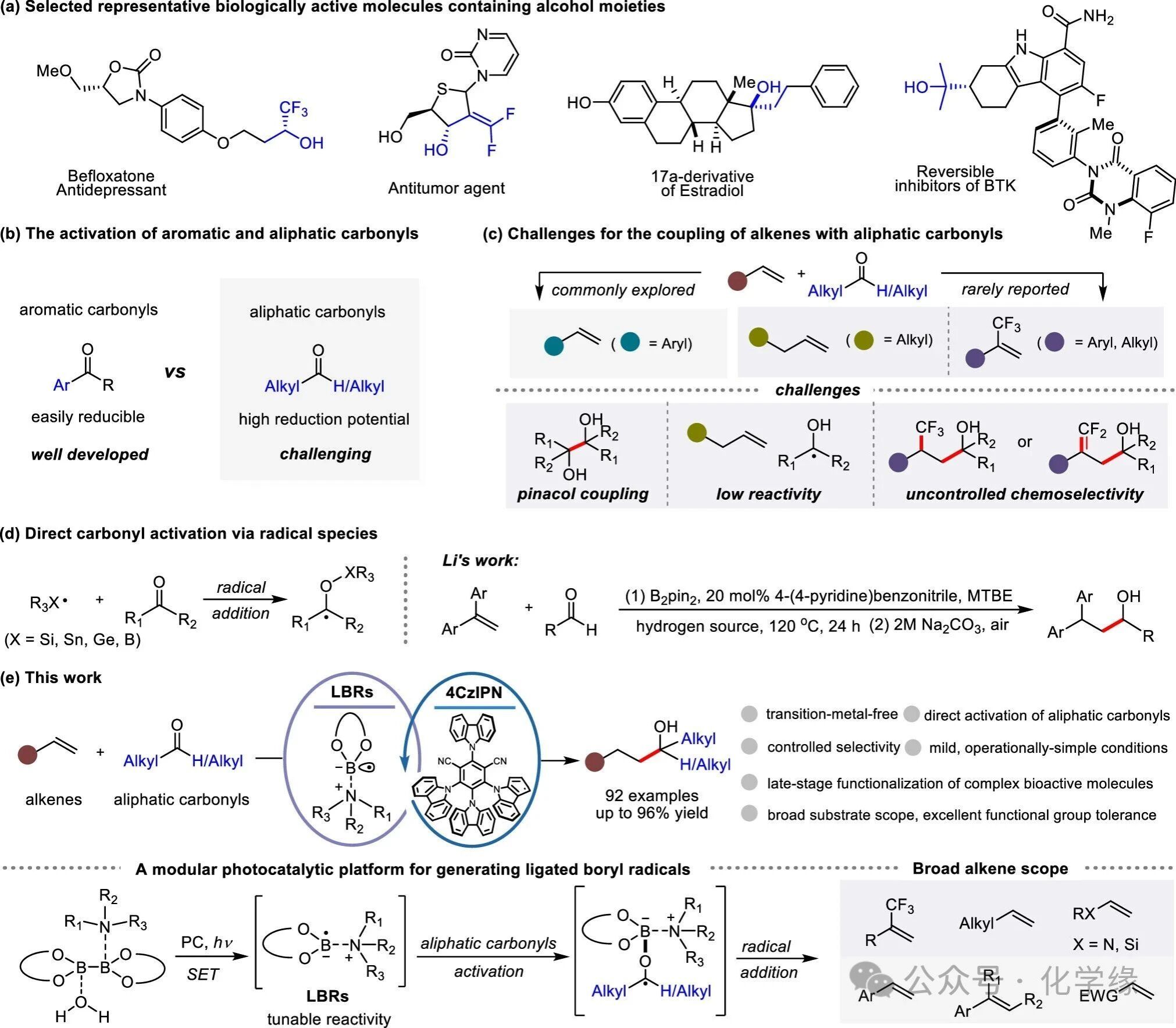
图 1 研究背景
羟基(−OH)是众多生物活性分子中的核心结构单元。其形成氢键与增强溶解度的特性,使其在分子识别、药理学及代谢过程中发挥着至关重要的作用,使含醇化合物成为生物医学领域不可或缺的存在。因此,开发高效合成此类结构的方法始终是合成化学领域的持续目标。在众多策略中,烯烃与羰基化合物的直接偶联因兼具商业可得性、稳定性和低成本优势而成为极具吸引力的简便途径,这一前提推动了近几十年的大量研究投入。尽管已取得显著进展,但相较于芳香族醛类,脂肪族醛基的直接活化仍面临持久挑战,主要源于其更高的还原电位及相关的热力学限制。
克服这一局限性的传统方法通常依赖于过量强金属还原剂,如锌、锰或二碘化钐。尽管有效,但与化学计量金属消耗相关的固有实际缺陷促使人们寻求替代催化策略。近期进展催生了强大的电化学和光化学方法,可直接活化脂肪族羰基与烯烃发生偶联反应。这些策略虽规避了计量金属还原剂的需求,却需依赖强还原性阴极或紫外光照来满足脂肪族羰基还原的高能耗要求,进而持续限制底物范围,导致可用的羰基化合物与烯烃种类受限。因此,现有方法主要聚焦于苯乙烯衍生物作为偶联单元,而其他烯烃(如脂肪族烯烃和α-三氟甲基烯烃)与脂肪族羰基化合物的偶联研究仍处于探索初期,成功案例极为有限。实现此类转化需解决若干关键挑战,包括抑制酮基自由基的竞争性频哪醇偶联反应、克服未活化烯烃的低反应性以及在使用α-CF₃烯烃时实现化学选择性控制。目前尚未有能同时应对所有这些挑战的通用催化策略被报道,这凸显了开发新型催化体系的迫切需求。
一种替代性的羰基活化策略利用自由基物种,包括有机硅基、有机锡基、有机锗基和硼基自由基,这些自由基可与羰基氧原子加成形成酮基自由基。其中,利用硼自由基生成交叉偶联反应活性中间体的策略备受关注。硼自由基已被证实能有效直接活化脂肪族羰基,实现酮基-烯烃偶联反应。Li团队代表性工作表明:通过4-氰基苯基吡啶介导的协同催化,在热条件下由B2pin2生成的吡啶配位的硼自由基,能高效活化脂肪族醛类形成酮基自由基,这些自由基随后与1,1-二芳基乙烯发生还原偶联反应。这项开创性工作确立了配位硼自由基在羰基活化中的潜力,但其实际应用受限于需高温操作且烯烃适用范围狭窄。尽管存在这些限制,这些发现仍凸显了配位硼自由基作为直接活化脂肪族羰基关键中间体的价值。未来,开发温和方法生成多样化配位硼基自由基仍是突破这些局限的理想目标。近期,作者基于光氧化还原催化,构建了由市售胺类与二硼酯结合的反应体系,该方法可在温和条件下生成多样化的配位硼自由基用于反应筛选。基于此进展,本文成功将该光氧化还原平台应用于连接硼自由基反应,突破前述技术瓶颈,实现了烯烃与脂肪族羰基化合物的多向偶联反应。该反应可兼容多种烯烃偶联试剂,包括α-三氟甲基烯烃、脂肪族烯烃、杂原子取代烯烃、苯乙烯及缺电子烯烃,较现有方法取得显著突破。该方法具有多重关键优势:无金属参与且反应温和,对官能团具有优异兼容性,并可通过简单调整催化体系实现可调的化学选择性。通过系统性调整光催化框架来精确调控反应结果的能力,凸显了该光氧化还原平台的多功能性和可调性。
图 2 反应条件优化
作者以α-CF₃苯乙烯O1与丙酮的偶联反应作为模型反应展开研究。尽管若干文献成功实现该转化,但这些方法通常依赖过渡金属催化,并使用锌或锰等化学计量比金属还原剂。此外,强吸电子的CF₃基团带来了显著的选择性挑战。现有方法大多局限于脱氟烷基化途径,仅能获得偕二氟烯烃产物。而针对α-CF₃烯烃与脂肪族羰基化合物进行氢烷基化反应的选择性调控,至今仍鲜有探索。近期,杨团队报道了基于光氧化还原催化、膦介导的六氟丙酮水合物脱氧反应,实现了α-CF₃烯烃的羟基聚氟烷基化,并选择性生成羟基烷基化产物而非脱氟烷基化产物。另方面,Shen团队通过精确选择光催化剂,实现了对脱氟烷基化或羟基烷基化产物的化学选择性调控。该体系未直接活化脂肪族羰基化合物,而是采用α-硅烷醇作为底物,通过质子偶联电子转移(PCET)过程实现活化。
当将光氧化还原策略应用于该反应时,通过简单调整催化体系即可有效引导化学选择性向脱氟烷基化或羟烷基化产物倾斜。当采用含2.0 mol % 4CzIPN、20 mol % 二异丙基乙胺A1及1.5当量二硼酯B1的催化体系时,O1与丙酮在室温下经450 nm LED照射1.5小时即可高效完成还原烯丙基脱氟偶联反应,以93%收率获得偕二氟烯烃叔醇a1,展现出优异的化学选择性。在此体系中,丙酮同时兼具反应物与溶剂的双重功能。采用3.0当量丙酮溶于二氯乙烷(DCE)的体系,亦可获得a1,收率达78%。光催化剂筛选发现:用Ir(dFMeppy)₂(dtbbpy)PF₆、Ir(ppy)₃或Ru(bpy)₃(PF₆)₂替代4CzIPN会降低反应效率和化学选择性,而Mes-Acr-Me+ClO₄⁻仅产生痕量a1。对二硼酸酯B2-B5的进一步评估证实,B1是与A1和4CzIPN形成脱氟烷基化产物a1的最佳配体。这一结果源于B1中特有的儿茶酚配体,其共轭效应显著降低了氧原子向硼中心供电子的能力。该效应增强了相应配位硼基自由基的电子缺失特性,从而提升其亲电性(相较于B2-B5体系),最终促进了羰基活化。
还探究了胺催化剂A2-A5的影响,发现氮原子中心的电子密度对反应选择性具有显著影响。当使用电子不足的胺A2时,反应路径明显从脱氟化转向质子化,主要生成羟烷基化叔醇b1。通过系统优化,采用20 mol % A2、1.5当量B2及2.0 mol % 4CzIPN条件下,成功实现羟烷基化叔醇b1的专一生成(收率86%)。在DCE溶剂中使用3.0当量丙酮亦可获得b1(收率68%)。
此外,评估了若干Lewis碱配位硼烷(常作为光氧化还原条件下配位硼基自由基的前体),但证实其对脱氟烷基化和羟烷基化途径均无效,凸显了该可调光氧化还原平台的多功能性与独特反应性。该反应对空气具有中等耐受性,尽管收率有所降低(a1为71%,b1为52%)。对照实验证实了胺催化剂、二硼酯、光催化剂和光照在形成a1和b1中的关键作用。
图 3 二氟烷基化的底物范围
Standard conditions: α-CF3 olefinsO (0.2 mmol), aliphatic carbonyls C (0.6 mmol), 4CzIPN (2.0 mol %), A1 (20 mol %), B1 (1.5 equiv), DCE (3.0 mL), r.t., Ar atmosphere and 450 nm LEDs irradiation for 1-3 h.
在确定优化反应条件后,采用A1/B1/4CzIPN催化体系进行二氟烷基化反应合成偕二氟烯醇的底物范围。具有多种官能团的α-CF3苯乙烯(包括苯环上不同位置的供电子和吸电子取代基)均能很好地兼容该反应,以良好至优异的收率(65-96%)得到目标产物。该反应同样适用于具有不同芳香骨架的α-CF₃烯烃,如芴、芘、吲哚、二苯并呋喃和噻吩,对应产物收率达67–92%。源自复杂生物活性分子的α-CF₃烯烃(包括环丙酸和吉非贝齐)得以高效转化为目标产物,收率分别为68%和67%。此外,在先前报道的条件下难以反应的脂肪族α-CF₃烯烃,在本光催化体系中顺利完成了脱氟烷基化反应,分别以85%和78%的收率得到对应产物。
随后考察了该转化中脂肪族羰基偶联配体的普适性。一系列线性脂肪族酮类反应高效,以67%至81%的收率生成叔醇。环状酮类同样被证实为有效底物,以良好收率生成目标产物。该方法可将天然产物和药物中的酮类直接与α-CF₃烯烃偶联,以良好收率得到目标产物。此外,该方法成功将烷基羰基化合物的适用范围扩展至醛类,实现了仲醇的合成,收率达55-86%。为验证该方法的实用性,进行了克级反应,顺利获得a2,收率达66%。
图 4 氢烷基化的底物范围
Standard conditions: α-CF3 olefinsO (0.2 mmol), aliphatic carbonyls C (0.6 mmol), 4CzIPN (2.0 mol %), A2 (20 mol %), B2 (1.5 equiv), DCE (3.0 mL), r.t., Ar atmosphere and 450 nm LEDs irradiation for 5-12 h.
通过简单切换至A2/B2/4CzIPN催化体系,随后研究了氢烷基化反应的底物范围以合成三氟甲基醇。所有反应在此条件下均展现出优异的化学选择性。首先,一系列在苯环上带有供电子或吸电子基团的α-CF₃苯乙烯类化合物顺利转化,以良好至优异的收率(61%–95%)得到对应的三氟甲基醇。b1的向上合成以62%收率完成,彰显了该方法的实用价值。此外,各类含杂芳环的α-CF₃烯烃(包括吡咯衍生物、吡啶衍生物、咔唑衍生物及二苯并呋喃衍生物)均以73%至85%的收率高效转化为目标产物。该策略成功应用于复杂生物活性分子(如左旋丙氨酸和扎洛普芬)衍生的α-CF₃烯烃的后期官能团化,获得中等收率的相应产物。
随后扩展了对脂肪族酮类适用性的研究。氘代丙酮能有效地掺入α-CF3烯烃,以72%的收率得到氘代三氟甲基醇。当采用不对称脂肪族酮时,尽管立体选择性仍难以控制,但仍以良好的收率获得了对应产物,且具有优异的化学选择性。此外,多种直链与环状酮均能与该反应兼容,生成烷基化醇类产物,收率达62–83%。
图 5 其他烯烃的底物范围
Standard conditions: olefins O (0.2 mmol), aliphatic carbonyls C (0.6 mmol), 4CzIPN (2.0 mol %), A1 (20 mol %), B1 (1.5 equiv), DCE (3.0 mL), r.t., Ar atmosphere and 450 nm LEDs irradiation for 1-3 h.
该光氧化还原策略不仅能实现脂肪族羰基化合物与α-CF₃烯烃的化学选择性偶联,更能兼容多种其他烯烃,此为其独特优势。包括含杂芳环的苯乙烯类化合物在内,均能与脂肪族羰基化合物高效反应,以良好收率生成对应醇类。含杂原子取代基的烯烃同样适用,以53%至94%的收率得到产物。除单取代烯烃外,1,1-及1,2-二取代烯烃亦可作为适宜的偶联对象,以74%至86%的收率生成对应产物。此外,一系列缺电子丙烯酰胺亦表现出良好兼容性,以57-90%收率生成对应产物。该策略成功应用于未活化烯烃,这类底物在常规条件下通常难以反应。在添加钪三氟甲磺酸盐(Sc(OTf)₃)后,反应得以进行并以中等收率得到产物。这些结果共同彰显了该方法的广泛底物适用性与优异官能团耐受性。
图 6 机理研究
为阐明反应机理,开展了一系列机理研究。循环伏安法实验首先揭示了胺在硼-硼键活化中的作用。自由基捕获实验证实自由基中间体的参与。通过氘标记实验确定了水是烷基化产物中的氢源。
图 7 可能的机理
基于上述机理实验及此前对该光氧化还原体系的研究,提出了一种合理的反应机制。该循环始于胺、二硼酯与H₂O形成复合物D,该复合物通过还原淬灭激发态光催化剂4CzIPN*,生成自由基阳离子中间体I和还原态光催化剂4CzIPN–。随后Int I中B-B键的断裂生成关键配位硼酰自由基Int II。该自由基与脂肪族羰基反应形成羰基自由基Int III,后者进一步攻击烯烃的C-C双键生成碳中心自由基Int IV。4CzIPN-对Int IV的单电子还原可再生光催化循环,并生成碳负离子Int V。最终,Int V经脱氟或质子化反应,分别形成脱氟烷基化或羟烷基化醇类产物。
总结:开发了一种通用且稳健的光氧化还原策略,通过硼自由基介导的烯烃与脂肪族羰基化合物的偶联反应,实现醇类化合物的发散性合成。该方法具有显著的底物广谱性,可处理α-三氟甲基烯烃、脂肪族烯烃、杂原子取代烯烃、苯乙烯及电子不足型烯烃,从而突破了先前方法的局限性。该策略的可调性通过简单改性催化体系即可实现对脂肪族羰基化合物与α-CF₃烯烃间精确的化学选择性调控,可选择性获得对映二氟烯醇或三氟甲基醇。此外,该无过渡金属反应体系可在温和条件下运行,无需传统方法常用的化学计量金属还原剂或强还原环境。其展现出优异的功能基耐受性,适用于复杂生物活性分子的后期官能团修饰。
文章信息:
A General Photoredox Platform for Divergent Couplings of Alkenes with Aliphatic Carbonyls via Ligated Boryl Radicals
Ting-Ting Miao, Rong-Bin Liang, Yao-Jun Wang, Xiang-Rui Li, Yonghong Xiao, Qing-Xiao Tong, and Jian-Ji Zhong*
DOI: 10.1021/acscatal.5c06961





